
Part 1: the paper with eight segments and eight co-first authors
Published:
Background
As mentioned in the introductory post this is the blog post about the Batson et al. (2021) study that started it all. When I joined Biohub remotely in late 2018 the California mosquito transcriptomes were still quite fresh but what do we do with these data? The main question that these pilot data were meant to be addressing - whether we can find viruses relevant to human health in mosquitoes prior to human cases - was clearly a “no”. What do we do now then? I spent much of 2018 and early 2019 probing these data and discussing potential avenues of research with the Biohub team - almost always Amy Kistler, Joshua Batson, Hanna Retallack, Lucy Li, and Kalani Ratnasiri - over evening Zoom calls (thanks to unfortunate differences between European and US West Coast timezones). Mind you these were pre-pandemic Zoom meetings, i.e. before it was cool. As usual, here’s the story of how the paper came about (from my perspective).
The power of repeating repeating repeating
As expected, and as will be discussed in another blog post, the more people focus on sequencing particular groups of hosts, the more likely they are to dredge up similar viruses. Though undeniably vast, virus diversity is finite after all. That does not mean that every project setting out to discover new viruses should be disappointed when previously described viruses are found. I think these cases help us out in two ways - first, multiple detections of the same genetic material acts as repetition code repetition code, an error correction method in coding theory where transmission of a message over a noisy channel achieves fidelity by repeating the message. Let’s say you assembled a contig with two completely overlapping ORFs running in opposite directions - was that a statistical fluke? It becomes difficult to argue that it is if someone else saw that before and even more so if your data has 40-odd contigs that look that way. Likewise for ORF positions, likewise contig ends, assembly artefacts, etc.
The second thing repeated detections of the same virus can do is something I’ve dreamed of for a while. Here’s the problem - your de novo assembled contigs from a metagenomic sequencing run is contig soup. Typically you’ll determine what’s what by sequence similarity to something that’s already known on public databases. If you have an RNA virus with an unsegmented genome you’re sorted - genes B, C, and D are clearly viral in origin even if they don’t resemble anything out there if they’re on the same piece of RNA with gene A that is a conserved virus gene that has annotated relatives. But good luck recognising this if genes B, C, and D are on separate segments of a segmented genome - they’re metagenomic “dark matter” now, genetic material you’d like to know about but cannot. There’s two approaches you can take here these days. If you work on a host that uses RNAi as its defense (like an arthropod) you can use small interfering RNA (siRNA) sequencing since those siRNAs will have been generated from double-stranded RNA (dsRNA) that was inside the cell and got caught. When not being used to regulate gene expression, dsRNA is also a dead giveaway of a replicating RNA virus so if you see siRNA reads aligning to your contigs it means your arthropod thought those contigs existed as dsRNA inside the cell and are strong virus candidates. Galbut (technically Galbūt) and Chaq viruses were discovered in this way by Darren Obbard and both names indicate uncertainty in Lithuanian and Klingon, respectively (two fictional languages, per Darren’s jest), since it was not 100% guaranteed they were actually viruses.
The other method for detecting likely viral sequences is simpler and gains power with each new dataset and relies on co-occurrence. Darren Obbard used this method to identify an entirely new family of RNA viruses in August 2019 and funnily enough Joshua Batson was writing code to do exactly the same thing around the same time. Back in January 2019 when presenting to the rest of the team I highlighted two viruses - Wuhan mosquito virus 6 (WuMV-6, an orthomyxo) and Culex narnavirus 1 (CxNV-1, a narna). WuMV-6 caught my attention because it was an orthomyxovirus that was seen before and quite common in our data while CxNV-1 had the unusual property of being ambigrammatic (having two completely overlapping ORFs running in opposite directions). In those January 2019 slides I put a rhetorical “fish out the rest of the genome?” near some early four segment (the most conserved PB1, PB2, PA, and NP) WuMV-6 phylogenies. When I took Josh’s code for a spin on our data my mind was immediately blown. By that point we already knew from Mang Shi and Eddie Holmes’ work that WuMV-6 had at least six segments but Josh’s code found another two. Obviously, they don’t resemble anything on GenBank and so could never be fished out of the contig soup by sequence similarity before but could be now - especially from other people’s data which convinced us we were seeing genuine segments. By the way - Culex narnavirus 1 turned out to have a previously unrecognised segment. Also ambigrammatic but more on that in a sister blog post.
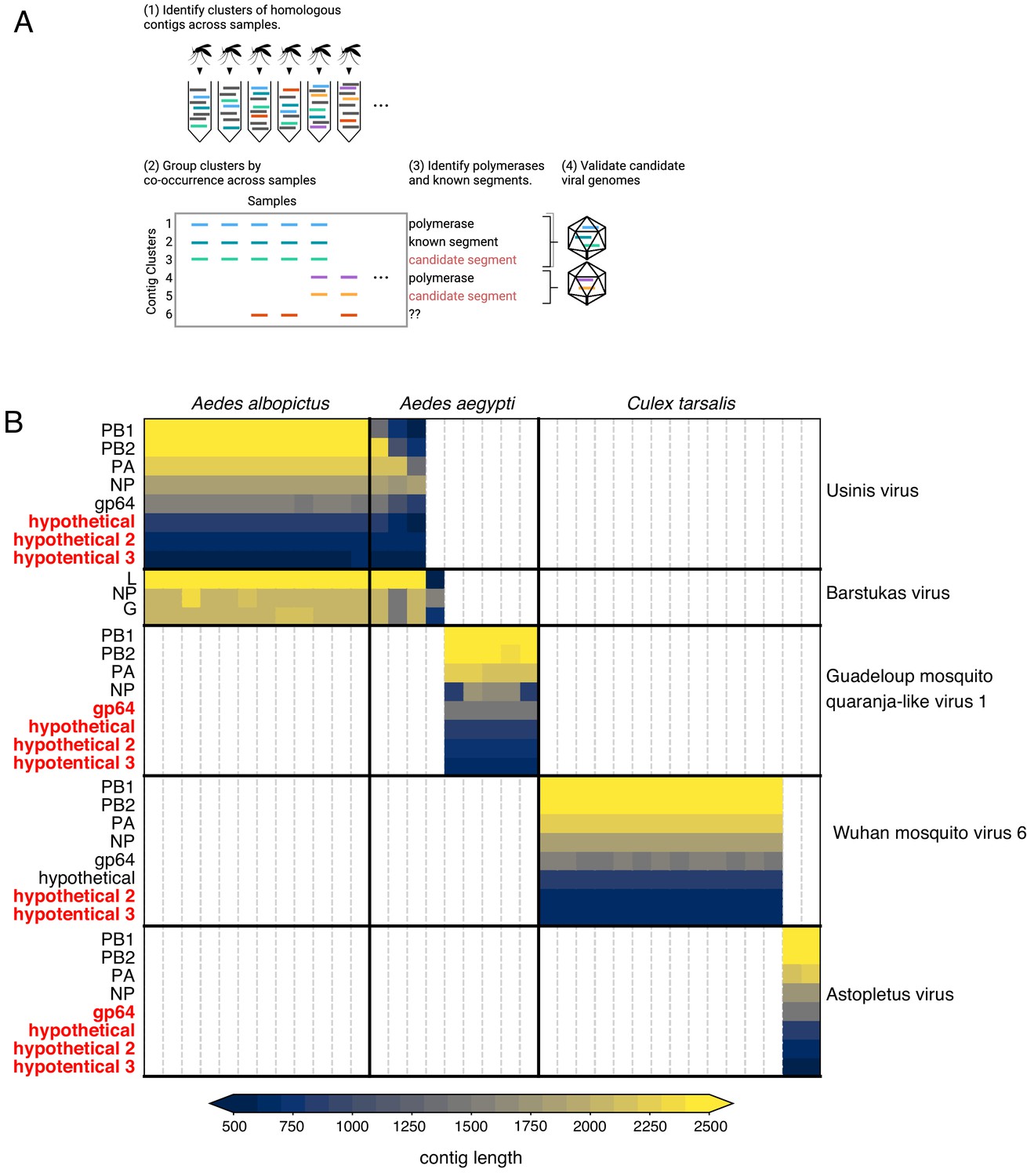
What now? Organise, re-organise
Armed with Josh’s new contig co-occurrence detection code Josh I realised we had to organise the viral portion of our data in a way that made sense moving forwards. Before we were relying on similarity (BLAST hits) to identify viral sequences and pull them out individually for other kinds of analyses (e.g. phylogenetics) or using BLAST results to look at the taxonomic breakdown of read counts. This wasn’t at all helpful in adding the metagenomic “dark matter” we pulled into the light with Josh’s code nor helpful in eventually submitting these sequences to GenBank. Josh’s code was quite helpful in another way - arthropod genomes can contain endogenous viral elements (EVEs) that are actually expressed as RNA so even if you identify a clearly viral contig in your transcriptome it’s not always guaranteed to be from a virus that’s there right now. We definitely had a few (-)ssRNA NP sequences hanging out in a few samples on their own nor co-occurring with anything else across multiple samples. Since arthropod EVEs (endogenous viral elements) are thought to come from viral mRNAs (where common transcripts are more likely to integrate) and since we were going to have to screen all of our contigs for co-occurrence, I reorganised all of our viral data based on RNA-dependent RNA polymerases (RdRps) since that’s a core requirement for an autonomous RNA virus and one of the rarer transcripts during infection that’s less likely to end up as an EVE. First we used hidden Markov models (HMMs) to fish out RdRps from our data then we used the RdRps as baits in Josh’s code to find the rest of the genome if it was segmented.
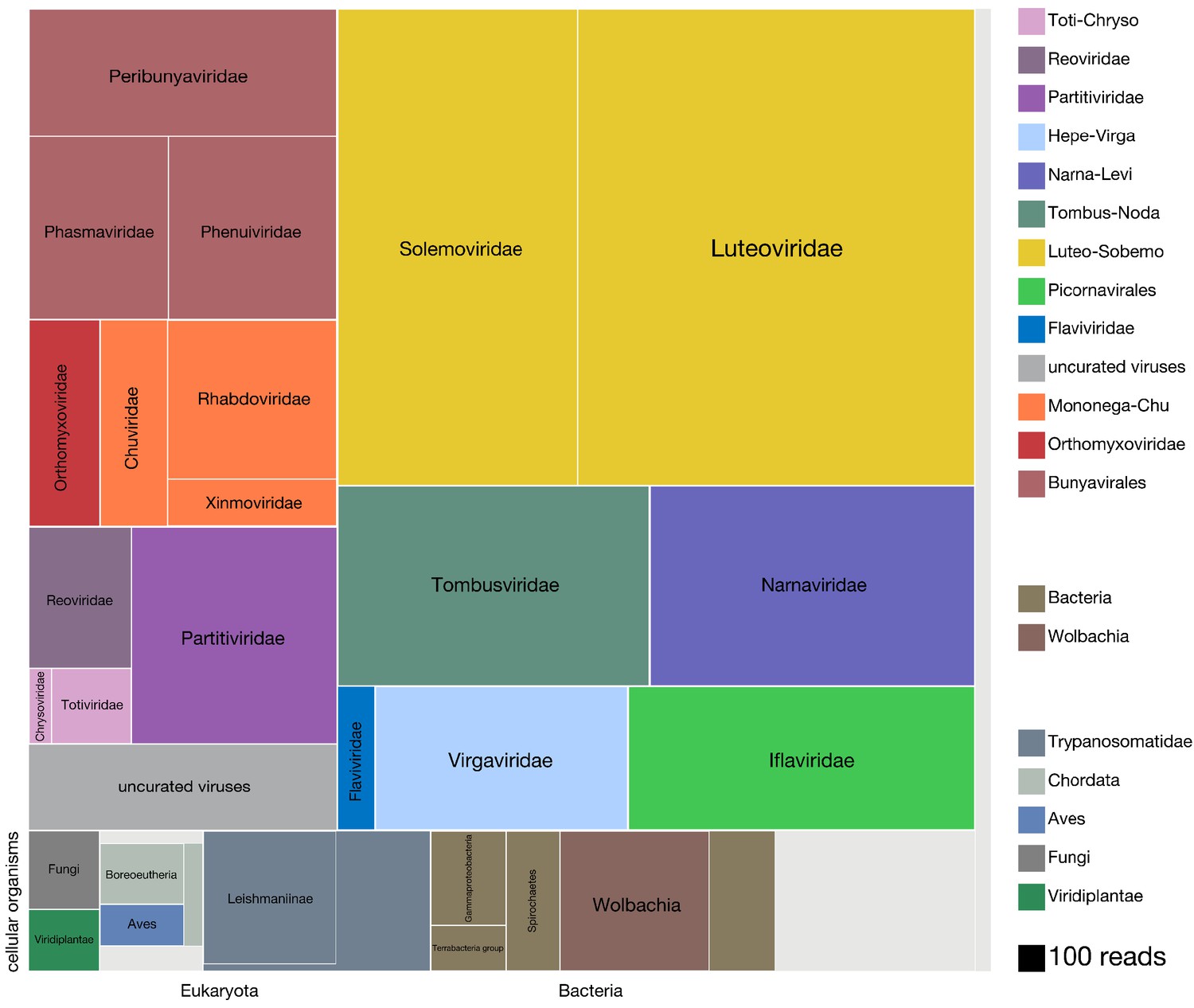
Over the course of this project another less pleasant reorganisation was happening on GenBank. Relatively early in my work on the treemap figure NCBI’s virus taxonomy (which I was using to organise the treemap compartments) suddenly changed to reflect one proposed alternative system, despite some very serious issues brought up about its methodology. In my opinion taxonomy is a fun little exercise that’s not to be taken too seriously since it’s subjective. On occasion virus taxonomy has made sense - Orthomyxoviridae, being a clade of (-)ssRNA viruses that use a heterotrimeric RdRp, and code for NP, at least one surface protein, and something to form a matrix, make sense as a taxonomic grouping, I’ll go as far as saying that the taxonomic rank of family might be useful for classifying viruses. However, in its current state virus taxonomy is peppered with ridiculous (read: over-split) and hypothetical (read: ambitious, given the divergence) taxonomic ranks that, if ever confirmed by research in the coming decades, I suspect will have been arrived at by chance. Annoyingly, following the transition to the new taxonomy I had viruses sitting both in the new (apparently) phylum Negarnaviricota (I’m fine with this since (-)ssRNA viruses are distinctly related) and the old “ssRNA negative-strand viruses”. Far from ideal. I guess the lesson from this little detour is that sometimes there will be people who care about something even if you don’t, so if you rely on things others have a say in you might be in for an unpleasant surprise down the line.
{kind=link}
Is it really a segment?
Using Josh’s co-occurrence method I was able to identify a total 15 novel segments associated with six viral RdRps and add more weight to the developing thought that Darren’s Chaq “virus” is actually a satellite segment of (also Darren’s) Galbut virus by observing co-occurrence of sequences related to both Chaq and Galbut (called Nefer virus in our data). Most (13) of the novel (i.e. unBLASTable) segments we identified belonged to orthomyxos - some were highly diverged surface proteins gp64 (but recognisable as such by HMM), and the rest (with the exception of WuMV-6 hypothetical segment discovered previously) were three small segments. It’s not surprising that we found the most new segments for orthomyxos since they have the highest recorded segment number amongst (-)ssRNA viruses and I’m convinced we missed segments of a couple of reoviruses in our data too (for the same reason).
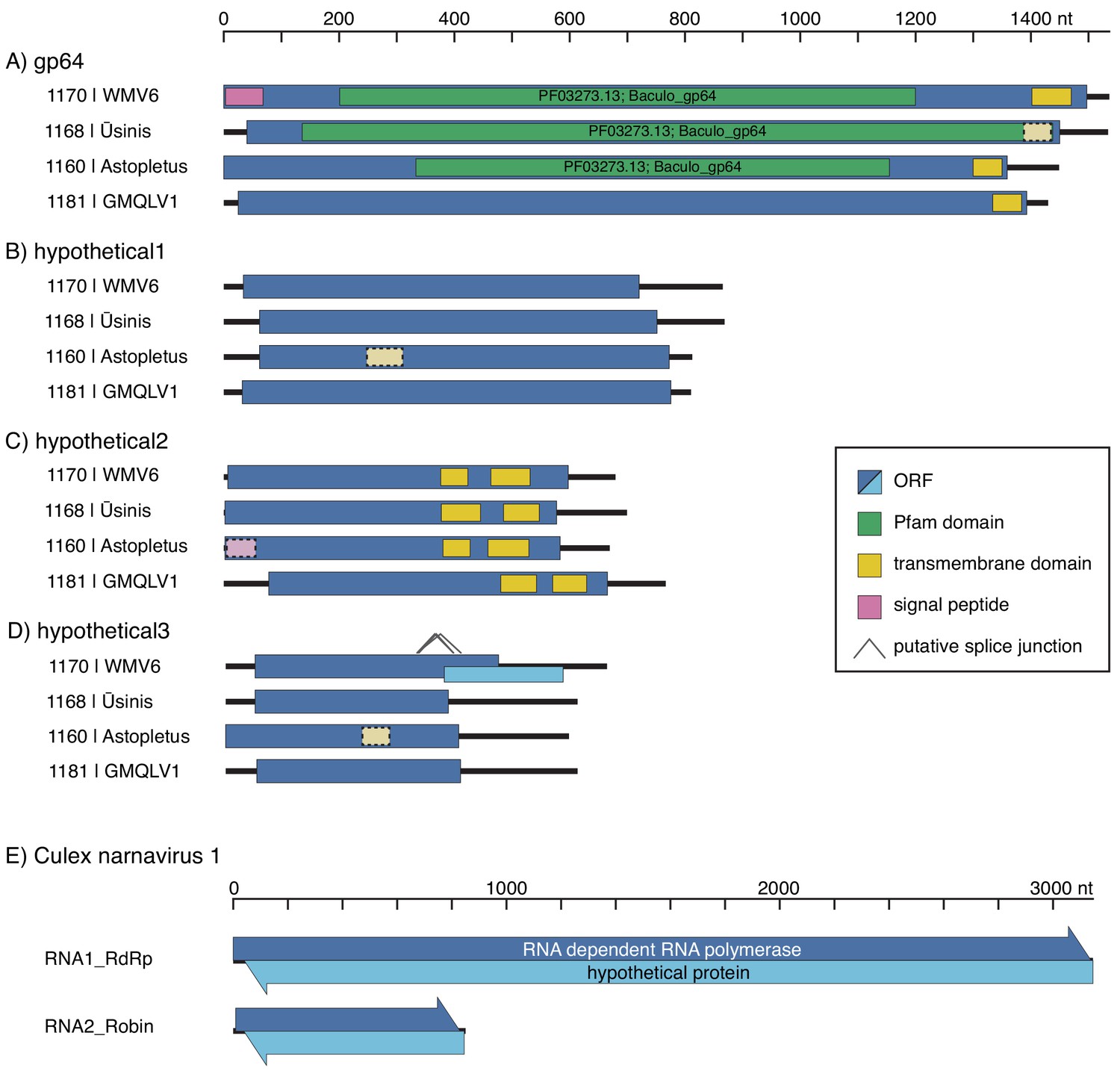
As we quickly discovered we couldn’t delve very deep into the new orthomyxo segments we found since not only did they not resemble anything on GenBank at that point but they didn’t even look like each other. Being the scientists that we are we therefore had to convince ourselves these segments were genuine and this was made quite easy: 1) putatively homologous segments shared features - length, transmembrane domains and their number in the case of segment hypothetical 2 or the longest ORF not occupying the full length of the segment and therefore likely to undergo splicing for segment hypothetical 3, 2) the phylogenetic trees we made for all segments of WuMV-6 were quite concordant across putative segments, and 3) we could either assemble all of the missing segments from other people’s data (for WuMV-6 and Guadeloupe mosquito-associated quaranjavirus 1) or saw reads that upon translation looked like they could be related, basically strong evidence that a broader clade of orthomyxos had this eight segment genome organisation. In my opinion, the ability to reconstruct these segments from other people’s data makes it conclusive and it’s very nice to see a proliferation of tools targeting the vast mountain of data sitting on NCBI’s sequence read archive (SRA) that help immensely with this task. Pebblescount and Serratus are the ones I know/use most, by the way.

The appearance of these tools is also a sign that many crews running sequencing experiments out there aren’t squeezing everything out of their raw data and why should they? Sequencing is ever cheaper and scientific questions motivating the sequencing are not always to do with virus discovery. Having said that, when reads come from virus discovery studies but only the most conserved / easiest to recognise contigs from segmented virus groups are submitted to GenBank I get a bit miffed. RdRps in their ever bewildering forms are extremely informative about RNA virus evolution but the rest of the genome is important too. Surface proteins come to mind here since something that’s receptor binding can (theoretically) make the difference between a virus that’s bloodborne or airborne or a virus that’s infecting you or a Trypanosome inside you. Obviously I’m more than happy to sort through the scraps if it means we’ll understand orthomyxo genome evolution a bit better and at the end of the day having mountains of sequence data that’s not been completely wrung out means there’s an enrichment of the research ecosystem for everyone else, particularly in resource-limited settings.
Fascinating virus ecology
Finally, there’s a couple of very interesting ways we broke down the distribution of viruses found in individual Californian mosquitoes. One obvious breakdown is to look at co-infection - how many distinct viruses do all of our sampled mosquitoes carry simultaneously? What does that histogram look like? We actually had nine mosquitoes without any viruses and one that had 13, the mode was three. The extremes were very telling too - samples without any viruses had low numbers of reads overall (so probably low quality) while six of the 13 viruses in the unfortunate mosquito were botourmiaviruses (thought to infect fungi) and probably came from the ergot fungus whose reads we also saw in the sample. The inability to determine the host of a given virus in metagenomic data is a known issue with some tools to address it (e.g. siRNA sequencing mentioned earlier) though I suspect with ever increasing amounts of sequence data we’ll come to rely more and more on the phylogenetic signal of certain virus groups associating with certain host groups as a useful proxy.
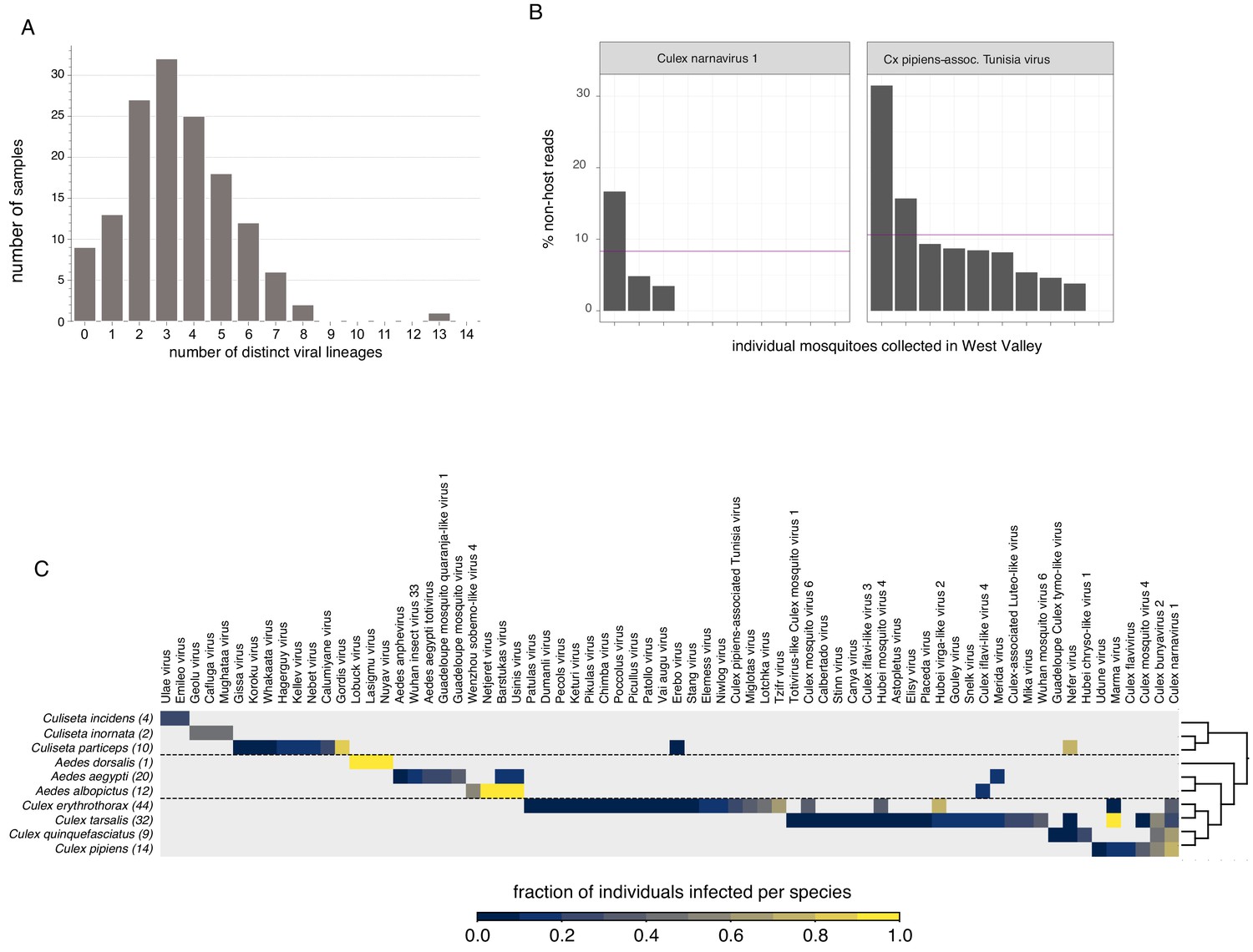
The next bit surprised me and I’m still not sure if it should have or not. At one point we decided to look at the distribution of viruses we found per mosquito species (bottom heatmap). Somewhat surprisingly, most of our viruses looked like they were very species-specific, e.g. Ūsinis is overwhelmingly found in Aedes albopictus (I suspect its presence in Aedes aegypti might be contamination) while Wuhan mosquito virus 6 (its closest relative) is found in Culex tarsalis. Having assembled numerous WuMV-6 genomes from SRAs it’s clear that WuMV-6 can also infect Culex quinquefasciatus, Cx. pipiens, Cx. australicus, and Cx. globocoxitus (Haemagogus, a member of Aedini too, allegedly). So on one hand we have a virus that’s not terribly picky about hosts as long as their Latin binomial starts with Culex but on the other hand we found it solely in Cx. tarsalis in California even though other hosts were available. This implies some sort of dynamics that we won’t be able to disentangle until we have more WuMV-6 genomes collected more consistently in time and space. The poor virome overlap between species also for the time being implies that many viruses might not be suitable for reconstructing community interaction networks because of sparseness.
In the shadow of the pandemic
Picture this timeline: in late 2017 the individual Californian mosquito transcriptomes are sequenced, in late 2018 I join the project, then in late 2019 I’m in SF and we’re finalising the manuscript until suddenly in early 2020 the SARS-CoV-2 pandemic begins. By this point we have a manuscript together. It’s a bit all over the place but there’s just that much interesting stuff in the sequences so we submit it to a journal as is. We get desk rejected, mostly because the manuscript isn’t focused enough but the editor adds insult to injury. In their opinion us highlighting Wuhan mosquito virus 6, a virus unrelated to the SARS-CoV-2 pandemic is inappropriate. That would be a fair comment if we didn’t have Github commits specifically mentioning WuMV-6 going back a year, i.e. before Wuhan was a household name. Unfortunately, this wasn’t the last or the most serious way the pandemic messed with this paper.
CZ Biohub and my co-authors ended up getting heavily involved in California’s SARS-CoV-2 surveillance efforts and that rejection in early 2020 meant that no one had any more time to revisit the manuscript until late 2020. Scrounging what little time we had between ourselves (and overwhelmingly thanks to Amy Kistler) we had a reworked draft ready in December 2020 and we decided to try out Review Commons which is basically outsourced peer review (and why not, reviewers aren’t being paid anyway). (Many things were happening then - the other two papers that I’ll discuss in sister posts A and B were born during this time, as well as Lithuania’s SARS-CoV-2 genomic surveillance programme.) We had the reviews from Review Commons back by February 2021 which were mostly positive so after another month of working on the comments during the precious hours between the mountain of work that was COVID-19 we sent the revised manuscript back to Review Commons. With the reviewers’ final approval we then had to decide which journal to submit to. Since we had positive reviews and a largely overhauled manuscript, I pushed Amy to submit to the same journal that desk-rejected us a year ago and to our surprise it got accepted! The rest is history.
What of all this?
To this day the individual California mosquito metatranscriptome project remains my favourite for many reasons. First and foremost, I got a chance to meet and work together with knowledgeable and lovely people (special shout out here to Joshua Batson, Amy Kistler, Lucy Lu, and Hanna Retallack) as a domain expert and working with experts from other domains. Don’t get me wrong, working on large genomic epidemiology studies was fun but it’s quite difficult to shine when everyone in the room has largely the same expertise as you.
Working on this study was also very rewarding because it allowed to make significant contributions to the orthomyxovirus field - fishing out novel segments that don’t resemble anything at all was a dream of mine for a long time. To be fair just working in metagenomics and virus discovery was a dream too. Finally, I have to thank this study for where I am today - by finding Wuhan mosquito virus 6 and the broader clade it belongs to I had a promising study system - not only accessible (WuMV-6 really does look like it’s everywhere) but also interesting in many fascinating ways. EMBO thought so too, we even talked a bit about it recently. Five years after joining a Zoom call with unfamiliar people sitting in San Francisco, today those people are still my friends and colleagues, I have more confidence in my abilities (see a theme?), and this whole academia thing seems like it’s going as well as it can.