
A VOC that wasn’t or SARS-CoV-2 lineage B.1.620 comes to Lithuania
Published:
You might’ve heard the unofficial motto of the SARS-CoV-2 pandemic - no one is safe until everyone is - floating around. Folks working in infectious disease understand it viscerally but I’m sure many outside the field still think it’s just some hippy slogan. So in this blog post I will tell you the story of SARS-CoV-2 lineage B.1.620 that landed on my lap thanks to a bit of (un)luck and a whole lot of globalisation. I figured that what transpired with lineage B.1.620 was somewhat unique and serves as a blunt statement about the planet we find ourselves on and how our failure to acknowledge that leads to a continuing cycle of avoidable mistakes. So let’s start at the beginning.
SARS-CoV-2 sequencing in Lithuania
For the first year of the pandemic I felt useless as I watched the UK and other European countries deploy routine sequencing as part of their response while all of the sequencing happening in Lithuania for a long time was one-off snapshots done at the initiative of individual academic groups. Like many countries in Europe, Lithuania didn’t have a SARS-CoV-2 genomic programme in place when the world first learned of B.1.1.7/Alpha in late 2020. When our government finally understood what they were missing out on I got the opportunity to write the SARS-CoV-2 genomic surveillance programme together with the fantastic Ingrida Olendraitė, Dovilė Juozapaitė, Daniel Naumovas, and Rimvydas Norvilas (more on them later). It was hard, thankless, and unpaid work with unreasonable hours, but I’m reasonably convinced no one else could have delivered this project this well. Regrettably, unwarranted institutional politics played out over those weeks during which I thought about quitting the project in disgust several times. Fortunately I didn’t and sequencing finally lifted off the ground in March 2021, with surveillance starting with samples from February 2021.
Deploying the sequencing resources of four local institutions and heavily relying on ECDC reference lab’s capacity Lithuania’s SARS-CoV-2 genomic surveillance programme has so far sequenced nearly 21,000 SARS-CoV-2 genomes, covering nearly 14% of all PCR-positive COVID-19 cases in Lithuania since February 2021. This continues to be a great source of pride to me personally and will be discussed in a separate blog post at some point in the future. The first thing we saw with the project was B.1.1.7’s transmission advantage play out over the next couple of months with the previous dominant B.1.1.280 and B.1.177.60 lineages being displaced by the interloper. It was looking like B.1.1.7 emerged as the final winner of the pandemic that would become the common ancestor of all future SARS-CoV-2 but of course that’s not what happened. But before Delta was even a thing in the midst of the rising B.1.1.7 wave in March-April due to lifting of restrictions in Lithuania we were suddenly faced with a pair of genomes that didn’t look right. The front-row seats to in-country lineage dynamics we got with the sequencing project were about to pay off.
Bad omens
The Sunday on April 11, 2021 started more or less typically - Ingrida Olendraitė posted assembled SARS-CoV-2 genomes and their lineage breakdowns to the Slack channel we’re using with Vilnius University Hospital Santaros Klinikos. “A boring run in terms of lineages - mostly B.1.1.7 and some B.1.177.60” she said. But that changed quickly, as Ingrida pointed out that a pair of genomes in the batch gave long branch warnings in nextclade. These two genomes - S21D420 and S21D421 - were classified by pangolin as B.1.177.57 but also had the (misleadingly sometimes called immune-evasive) S:E484K mutation. We knew S:E484K is picked up quite frequently by other lineages but the placement of these two genomes in nextclade made it clear that we weren’t dealing with anything close to B.1.177. Our mystery genomes were directly derived from a B.1-like genotype and them sitting on a long branch meant they had either been circulating somewhere undetected for a while or experienced similar selective pressures to B.1.1.7/Alpha.
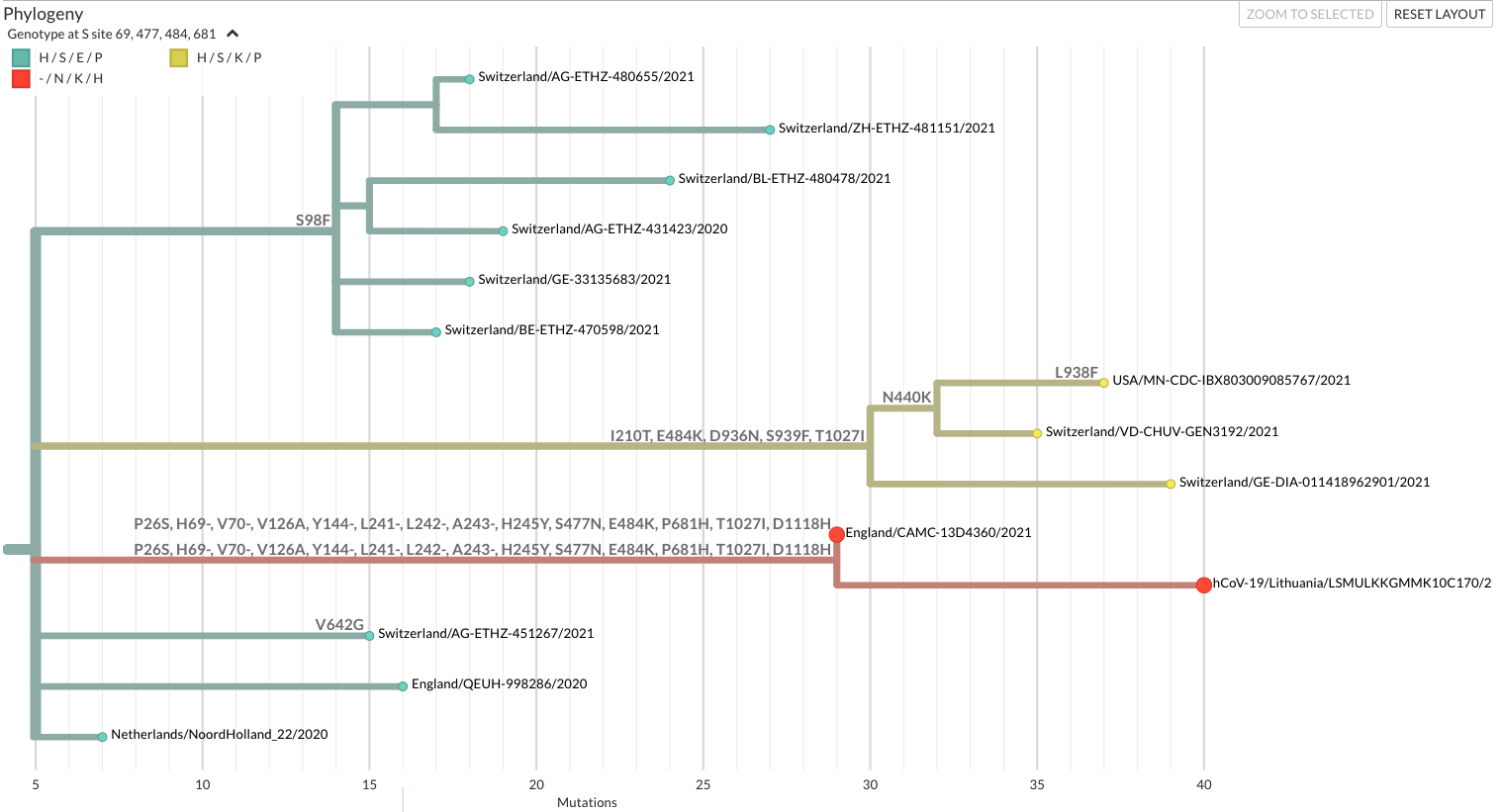
The incorrect pangolin classification of these two mystery genomes as B.1.177.57 when they clearly weren’t anywhere near B.1.177 meant it would be hard to find their relatives on GISAID and the worst thing about them were the actual mutations on the long branch they sat on. It was a smörgåsbord of mutations found in classic variants of concern of the day (Alpha, Beta, and Gamma) - P26S, 69/70Δ, 144Δ, 241/243Δ, S477N, E484K, P681H, and D1118H to name the S changes alone. It looked like a cartoon villain. The mutations seemed so excessive my first instinct was to check for contamination, but that didn’t seem likely since we had two genomes, not a singleton, and Betas and Gammas that could be the source of contamination weren’t circulating in Lithuania widely. Screwing up so consistently seemed a bit of a stretch. Was it recombinant? We already knew those existed thanks to Ben Jackson and co, but in this case we were missing a number of mutations that a B.1.1.7-derived recombinant should’ve had. So it probably wasn’t a recombinant either.
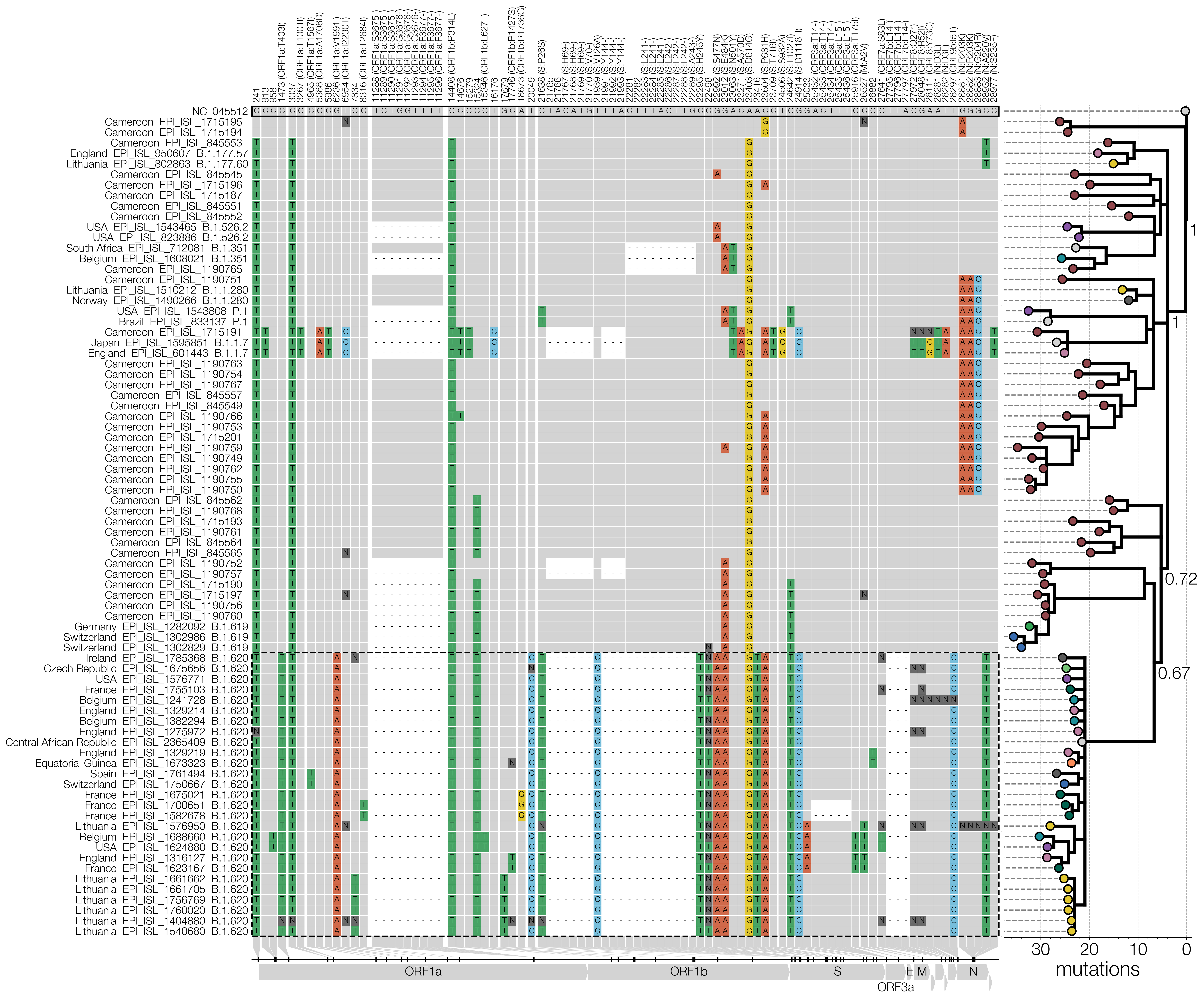
If we weren’t convinced by the two genomes we had at hand, we soon discovered that searching for the combination of 69/70Δ, S477N, and E484K mutations on GISAID lead to a very small (at the time) list of genomes that happened to carry the rest of the mutations too. Worse yet, that’s how we found out that another sequencing institution in Lithuania had seen this lineage a week earlier but took the pangolin B.1.177.57 label at face value and missed the S:E484K mutation. We informed government institutions to step up their game in the affected area (Anykščiai municipality) while we turned our attention to where this mystery lineage may have come from.
The other 30-odd genomes on GISAID with 69/70Δ, S477N, and E484K reported mid-April were from a pretty random collection of European countries: France, Belgium, Switzerland, England, and Germany, all of them collected no earlier than March 2021. An unusual lineage suddenly appearing across much of Europe was another red flag, kind of like early B.1.1.7/Alpha spread in Kent vibes. Often genomes of this lineage from the same country weren’t even closely related to each other so given the sudden appearance of these genomes across numerous European countries and that we couldn’t identify any genomes close enough to break up the super long branch leading up to these sequences led to our initial hypothesis that we were seeing something that was not endemic to Europe. Checking the data again we got our first confirmation that we were on the right track - the earliest case on GISAID was marked as a traveler returning to France from Cameroon. As luck would have it I knew the perfect group who could analyse traveler sequences in BEAST and I could certainly use more help.
Unhinged April
Guy Baele, Sam Hong and Barney Potter were a blessing for this project. Guy initially helped emailing folks who submitted genomes with 69/70Δ, S477N, and E484K to GISAID which by the time we were writing up had yielded a total of eight travel cases arriving from Cameroon. Sam Hong was crafting the BEAST XMLs we’d eventually use and sorted out background sequence data while Barney Potter ran analyses to look for sequences that could represent “missing links” in the evolution of our mystery lineage that could break up the long branch these genomes were sitting on. Simultaneous with these efforts we also submitted a pango lineage proposal, because listing the mutations this lineage had in every email to every new correspondent was getting tedious. To my surprise and perhaps with a lot of luck we got a pango lineage name the very next day and internally we no longer had to refer to the lineage as Puntukas or Anykščių šilelis in Lithuania.
What followed was a 23-day writing spree that resulted in the final paper. We settled on the general outline of the study fairly quickly since there’s not much room to reinvent the wheel in a study describing a new lineage. You begin by listing the mutations it has, show some trees, describe its range, and say some things about its potential future. I struggle to remember a similar time when I knew exactly what had to be done or a time when I could work on a single project undistracted for 12 hours per day for three weeks. I would never endorse it as a healthy long-term strategy, but going to bed tired because you’re practically obsessed, able to make good progress on a project and know exactly what needs doing tomorrow is kinda nice. But only in moderation, especially because on top of actual work I was thrust into deeply unpleasant PR territory that no one else could handle.
At one point Lithuanian media caught wind that we had found an “unidentified” coronavirus strain that I simply didn’t have time nor patience to explain to every media outlet in the country. So on I went, juggling press releases and analyses, coordinating data, and inviting co-authors while trying to ignore all the cringily awful headlines reporting on our work that verged on snakerona (raise your hand if you remember that story) and Ebola mutating OMG!!1!. Everyone wanted juicy scoops to write clickbait-y headlines with but no one bothered reading up on the subject matter. At one point a journalist confidently told me that “mutation” and “strain” are used as synonyms in the news and didn’t seem to know why they wanted to interview me specifically, just that there were rumours of something called “sequencing”. I wish more journalists everywhere treated their profession as a calling to knowledgably report information to society rather than just a means to collect a paycheck based on the headline clicks they get.
Visualisations
Even though the paper itself was quite formulaic it did require the development of new code for visualisations that I wanted to do. Figures 1 and 2 are the only ones worth discussing here since they required the most work. Anyone who’s been following SARS-CoV-2 closely will recognise our Figure 1 as a blatant clone of Áine O’Toole’s snipit visualisation with my sole contribution being the addition of a tree. I initially saw snipit in the first(?) report on genuine SARS-CoV-2 recombinants and while I’ve done condensed SNP alignments previously I didn’t appreciate that they could make for very informative and aesthetic figures.

The other thing that experts might find obvious but the novices will (hopefully) appreciate is connecting the tree to the data underlying it (the SNPs). My impression is that unless you’re working with Geneious or nowadays nextstrain/auspice, your tree-inference tool will not let you switch between the alignment and the tree or run ancestral state reconstruction, and so the tree is then somewhat divorced from its source data. I’ve seen ridiculously long branches in trees caused by misalignments and back-translated sequences that I spotted in alignments on time but I wonder how many beginner phylogeneticists not using tools that allow one to check the alignment easily would just proceed with the next steps that follow without looking back.
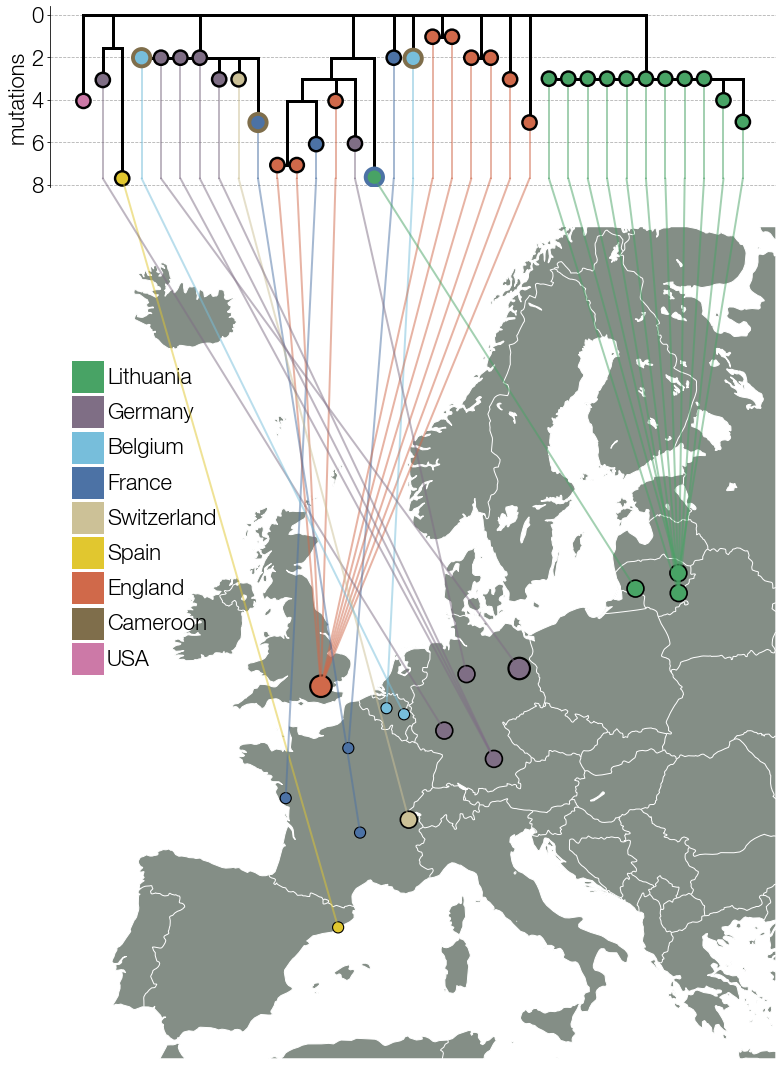
Figure 2 (from earlier) was very much a clone of Nídia Trovão’s figure literally connecting trees to geography which I wouldn’t know where to begin with in matplotlib until they implemented ConnectionPatch, an object that can span multiple matplotlib axes objects. When we initially had 30-odd B.1.620 genomes the figure was pretty much a carbon copy of Nídia’s.
{kind=link}
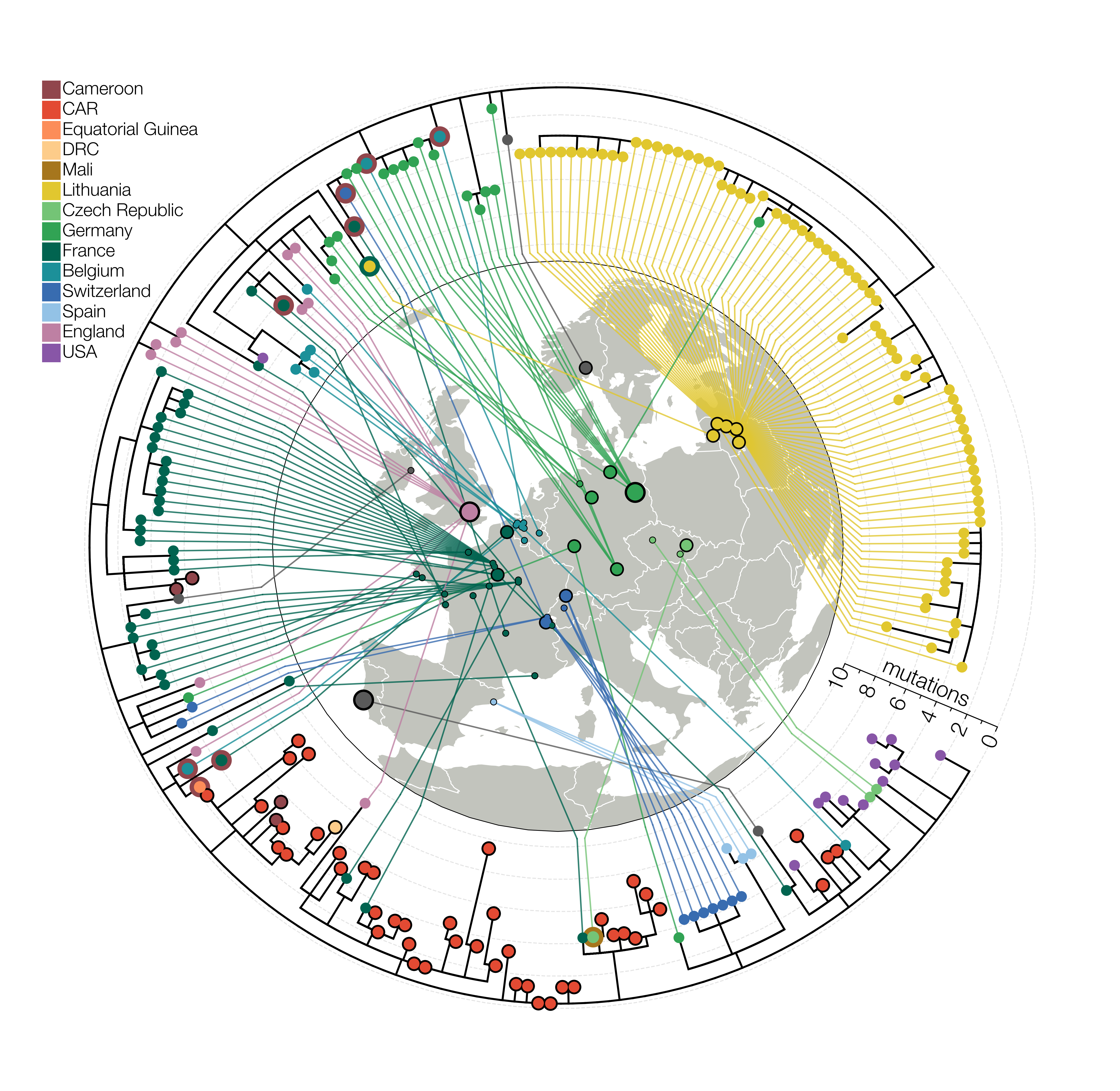
But at some point, as the number of genomes grew I quickly realised what I must do, I just didn’t know if I had the strength to do it. I had to go circular. It’s the one thing that myself and experienced phylogeneticists advise people not to do unless it’s for logos but I felt it was justified at the time because surrounding the map with a tree pointing inwards gave us the space we needed, it looked hip, the tree wasn’t so complicated as to be uninterpretable, it was perfect. Novel, crisp, and best of all - it told a story. But then the reviewers happened. One of the requests we received during peer-review was to include more genomes in case they changed the story. For the last six months the first thing I did in the morning was to go on GISAID to check for new B.1.620 genomes. I knew nothing new that affected our story in any way had come to light but obviously you don’t say that to reviewers. A couple of months after the initial submission there were loads more B.1.620 genomes with loads more sequencing/assembly errors which made the tree topology more complicated and the tree itself more crammed. I’m not a fan of the peer-reviewed Figure 2 we ended up with so to me Figure 2 lives in its perfect submitted form.
Much ado about nothing?
We didn’t get to see if B.1.620 was all that bad thanks to B.1.617.2/Delta. There were hints from the mutations it carried and its persistence that B.1.620 probably had an advantage over B.1.1.7/Alpha in vaccinated populations that could have led to a uniquely B.1.620-dominated 2021 winter season in Lithuania. It was designated as a variant under monitoring by the ECDC together with WHO-designated variants of interest B.1.621 (WHO designation Mu) and C.37 (WHO designation Lambda) but never got its own WHO VOI designation. But in many ways the B.1.620 story wasn’t even about the lineage itself but about the failures that led to it.
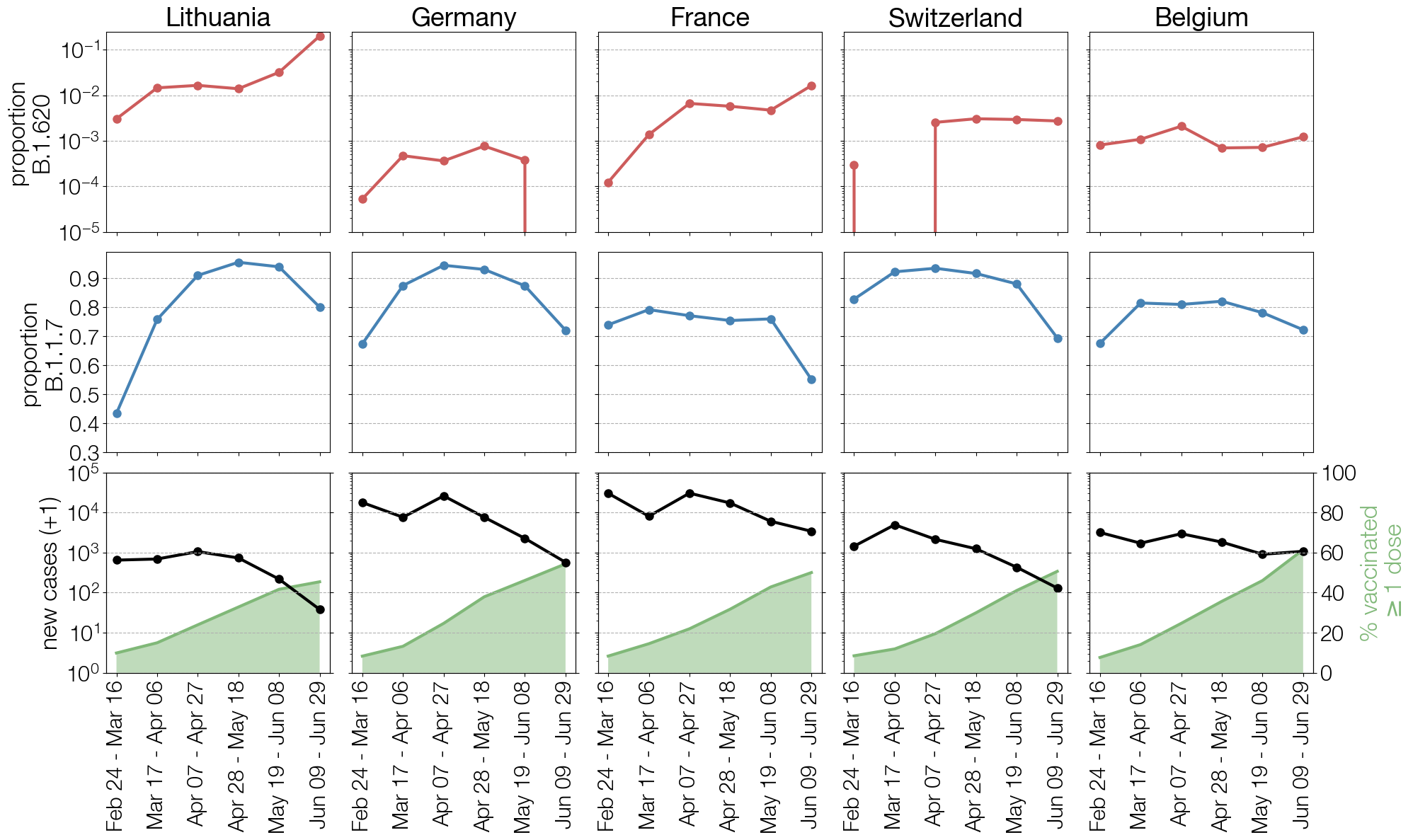
Locally, I believe we spotted B.1.620 comparatively early and the call to action by some in the government was met with lukewarm efforts within institutions responsible for enacting the response. I don’t know if it was because the people responsible weren’t being paid enough to care or if the Soviet spirit of doing the bare minimum to fly under the radar was still alive and well within the institution but by the end of B.1.620’s rampage we could point to nearly 200 B.1.620 cases spread across all of Lithuania confirmed by sequencing and over 400 strongly suspected cases based on genotyping as evidence of a botched response, since all of our B.1.620 cases descended from a small outbreak in a single municipality. I wouldn’t be surprised if the responsible institutions simply didn’t understand that their failure to act on time would be clear as day in sequence data. I’m also somewhat fascinated about another independent introduction of B.1.620 into Lithuania (yes, we somehow had the sheer unluck of having two introductions) which turned out to be a traveler who as far as we could tell from sequence data did not infect anyone upon their return. By some twist of fate not only did we have an outbreak of an unusual lineage in Lithuania but we also got the perfect example of how the decisions of single individuals can have a disproportionate impact on those around us in an infectious disease setting. One person decided to self-isolate responsibly and another probably didn’t and inadvertantly led to the deaths of a number of their compatriots.
On a more global scale even though B.1.620 is likely to be extinct, the wider lesson it teaches doesn’t need for B.1.620 to still be circulating. We often talk about global inequalities that rob poorer countries of opportunities but when we argue that it’s a problem we frequently have to rely on evidence from absence. Absence of comparable numbers of scientists, papers, institutions, etc coming from poorer countries. I think B.1.620 was an interesting twist on this argument. The lack of vaccines in central Africa probably allowed B.1.620 to evolve in the first place while the lack of sequencing there robbed richer countries of an early warning that B.1.620 even existed until it was knocking on our door. Everyone ended up losing and it didn’t matter that the Baltic states and central Africa are about as random as any two locations in the world can get. The de facto world we find ourselves living in is considerably smaller than it used to be, it has been for a while, and it gets smaller every year. We can continue pretending that problems elsewhere aren’t ours but reality will call this bluff every time, to our disadvantage.
The other dream team(s)
Although Guy, Sam and Barney did most of the heavy lifting for our B.1.620 study I would also like to acknowledge the folks whose work enabled us to do so in the first place. I have to thank Ingrida Olendraitė for getting me involved in Lithuania’s pandemic response at the end of 2020. Ingrida had been an irreplaceable left hand (#teamLeftie) in everything to do with Lithuania’s SARS-CoV-2 sequencing project while still writing up her thesis. It’s been an honour and a pleasure to work with such a promising young scientist and I’m already looking for ways to continue collaborating. When the government was finally ready to fund the sequencing project, writing the actual genomic surveillance project proposal could not have happened without the help of Rimvydas Norvilas, Dovilė Juozapaitė, and Daniel Naumovas. While Ingrida and I worked on the “why do this” and “what needs to happen” aspects of the project they worked out all of the technical details. When the sequence data were finally rolling in it quickly became apparent that government institutions needed a person who could clean the metadata. Luckily for us Miglė Gabrielaitė stepped up to the challenge and volunteered her time despite being in the final stages of her PhD and expecting. Aistis Šimaitis from the government’s chancellery and Jonas Bačelis from the government’s statistics department were crucial for coordinating the data infrastructure required to pull off a project with this amount of throughput and numerous other scientists working at Vilnius University Life Sciences Center, Lithuanian University of Health Sciences, Vilnius University Hospital Santaros Klinikos, and Lithuanian University of Health Sciences Hospital Kauno Klinikos also contributed the key ingredient to our study - Lithuanian SARS-CoV-2 genomes. Since this study came out I’ve moved back to Lithuania and I couldn’t have asked for a better way for the Lithuanian science community to get to know me and vice versa.
Though getting sequencing up and running in Lithuania was a challenge in many ways, some of the hurdles that inconvencienced us somewhat (and probably would’ve been easy to solve in richer countries) obviously had to present substantial challenges for groups sequencing in Africa. To date the closest relatives of lineage B.1.620 - its sibling lineage B.1.619 and intermediate precursor-looking lineages were found in Cameroon and were sequenced, as far as I can tell, because Ahidjo Ayouba took the initiative to do so. It highlights yet again that sequencing regularly, regardless of how boring the current state of the epidemic looks, can contextualise future sequences in unforeseen and informative ways. Similarly, as we were writing up the B.1.620 story, numerous groups around Africa were able to get their samples sequenced and submitted to GISAID, often with the help of INRB, who have time and again stepped up to do necessary sequencing in DRC (and abroad this time). I’m very glad we were able to add the names of researchers who sequenced B.1.620 in Africa to the paper to acknowledge their efforts.
A short note on impostor syndrome
Most scientists encounter impostor syndrome in one way or another at some point in their careers and I’m no exception. A superior can reassure us that we’re on the right track and discussing our work with colleagues helps us approach problems from a wider set of angles and get more minds working on the problem. Since leaving Seattle in the summer of 2018 I haven’t had the luxury of either but I managed to cope okay because I was working on RNA virus discovery projects proceeding at their own pace. SARS-CoV-2 work, on the other hand, required a degree of urgency and precision. My happiness at making predictions based on limited data might seem childish to the experienced phylogeneticist because in retrospect the initial observations were quite obvious, but trusting my own abilities to make inferences in the absence of superiors and colleagues telling me “yes, that makes sense” took a lot of effort. Though I’m nowhere near “cured” of my impostor syndrome at least I got some vaguely objective evidence for why I’m not completely useless.